Metagenome Assembly Workflow (v1.0.1)¶
Workflow Overview¶
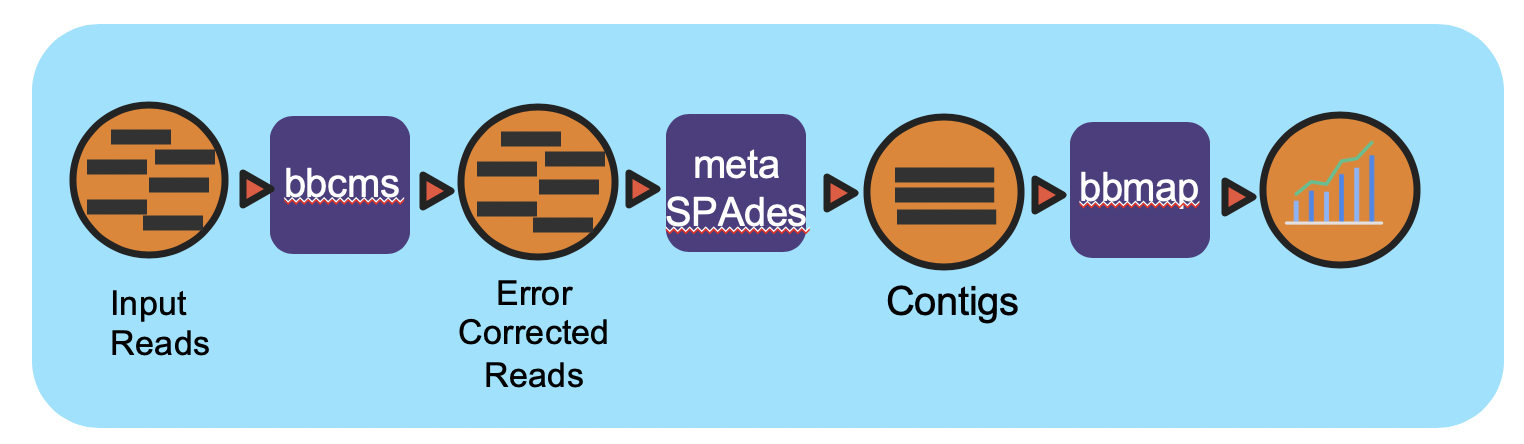
This workflow takes in paired-end Illumina reads in interleaved format and performs error correction, then reformats the interleaved file into two FASTQ files for downstream tasks using bbcms (BBTools). The corrected reads are assembled using metaSPAdes. After assembly, the reads are mapped back to contigs by bbmap (BBTools) for coverage information. The .wdl (Workflow Description Language) file includes five tasks, bbcms, assy, create_agp, read_mapping_pairs, and make_output.
- The bbcms task takes in interleaved FASTQ inputs and performs error correction and reformats the interleaved fastq into two output FASTQ files for paired-end reads for the next tasks.
- The assy task performs metaSPAdes assembly
- Contigs and Scaffolds (output of metaSPAdes) are consumed by the create_agp task to rename the FASTA header and generate an AGP format which describes the assembly
- The read_mapping_pairs task maps reads back to the final assembly to generate coverage information.
- The final make_output task adds all output files into the specified directory.
Workflow Availability¶
The workflow from GitHub uses all the listed docker images to run all third-party tools. The workflow is available in GitHub: https://github.com/microbiomedata/metaAssembly; the corresponding Docker images are available in DockerHub: https://hub.docker.com/r/microbiomedata/spades and https://hub.docker.com/r/microbiomedata/bbtools
Requirements for Execution¶
(recommendations are in bold)
- WDL-capable Workflow Execution Tool (Cromwell)
- Container Runtime that can load Docker images (Docker v2.1.0.3 or higher)
Hardware Requirements¶
- Memory: >40 GB RAM
The memory requirement depends on the input complexity. Here is a simple estimation equation for the memory required based on kmers in the input file:
predicted_mem = (kmers * 2.962e-08 + 1.630e+01) * 1.1 (in GB)
Note
The kmers variable for the equation above can be obtained using the kmercountmulti.sh script from BBTools.
kmercountmulti.sh -k=31 in=your.read.fq.gz
Workflow Dependencies¶
Third party software: (This is included in the Docker image.)¶
- metaSPades v3.15.0 (License: GPLv2)
- BBTools:38.90 (License: BSD-3-Clause-LBNL)
Sample dataset(s)¶
Zymobiomics mock-community DNA control (SRR7877884); this dataset is ~4 GB.
Note
If the input data is paired-end data, it must be in interleaved format. The following command will interleave the files, using the above dataset as an example:
paste <(zcat SRR7877884_1.fastq.gz | paste - - - -) <(zcat SRR7877884_2.fastq.gz | paste - - - -) | tr '\t' '\n' | gzip -c > SRR7877884-int.fastq.gz
For testing purposes and for the following examples, we used a 10% sub-sampling of the above dataset: (SRR7877884-int-0.1.fastq.gz). This dataset is already interleaved.
Input¶
A JSON file containing the following information:
- the path to the input FASTQ file (Illumina paired-end interleaved FASTQ) (recommended the output of the Reads QC workflow.)
- the contig prefix for the FASTA header
- the output path
- memory (optional) ex: “jgi_metaASM.memory”: “105G”
- threads (optional) ex: “jgi_metaASM.threads”: “16”
An example input JSON file is shown below:
{
"jgi_metaASM.input_file":["/path/to/SRR7877884-int-0.1.fastq.gz "],
"jgi_metaASM.rename_contig_prefix":"projectID",
"jgi_metaASM.outdir":"/path/to/ SRR7877884-int-0.1_assembly",
"jgi_metaASM.memory": "105G",
"jgi_metaASM.threads": "16"
}
Output¶
The output directory will contain four output sub-directories: bbcms, final_assembly, mapping and spades3. The main output, the assembled contigs, are in final_assembly/assembly.contigs.fasta.
Part of an example output JSON file is shown below:
├── bbcms
│ ├── berkeleylab-jgi-meta-60ade422cd4e
│ ├── counts.metadata.json
│ ├── input.corr.fastq.gz
│ ├── input.corr.left.fastq.gz
│ ├── input.corr.right.fastq.gz
│ ├── readlen.txt
│ └── unique31mer.txt
├── final_assembly
│ ├── assembly.agp
│ ├── assembly_contigs.fasta
│ ├── assembly_scaffolds.fasta
│ └── assembly_scaffolds.legend
├── mapping
│ ├── covstats.txt (mapping_stats.txt)
│ ├── pairedMapped.bam
│ ├── pairedMapped.sam.gz
│ ├── pairedMapped_sorted.bam
│ └── pairedMapped_sorted.bam.bai
└── spades3
├── assembly_graph.fastg
├── assembly_graph_with_scaffolds.gfa
├── contigs.fasta
├── contigs.paths
├── scaffolds.fasta
└── scaffolds.paths
The table provides all of the output directories, files, and their descriptions.
| Directory | File Name | Description |
|---|---|---|
| bbcms | Error correction result directory | |
| bbcms/berkeleylab-jgi-meta-60ade422cd4e | directory containing checking resource script | |
| bbcms/ | counts.metadata.json | bbcms commands and summary statistics in JSON format |
| bbcms/ | input.corr.fastq.gz | error corrected reads in interleaved format. |
| bbcms/ | input.corr.left.fastq.gz | error corrected forward reads |
| bbcms/ | input.corr.right.fastq.gz | error corrected reverse reads |
| bbcms/ | rc | cromwell script sbumit return code |
| bbcms/ | readlen.txt | error corrected reads statistics |
| bbcms/ | resources.log | resource checking log |
| bbcms/ | script | Task run commands |
| bbcms/ | script.background | Bash script to run script.submit |
| bbcms/ | script.submit | cromwell submit commands |
| bbcms/ | stderr | standard error where task writes error message to |
| bbcms/ | stderr.background | standard error where bash script writes error message to |
| bbcms/ | stderr.log | standard error from bbcms command |
| bbcms/ | stdout | standard output where task writes error message to |
| bbcms/ | stdout.background | standard output where bash script writes error message(s) |
| bbcms/ | stdout.log | standard output from bbcms command |
| bbcms/ | unique31mer.txt | the count of unique kmer, K=31 |
| spades3 | metaSPAdes assembly result directory | |
| spades3/K33 | directory containing intermediate files from the run with K=33 | |
| spades3/K55 | directory containing intermediate files from the run with K=55 | |
| spades3/K77 | directory containing intermediate files from the run with K=77 | |
| spades3/K99 | directory containing intermediate files from the run with K=99 | |
| spades3/K127 | directory containing intermediate files from the run with K=127 | |
| spades3/misc | directory containing miscellaneous files | |
| spades3/tmp | directory for temp files | |
| spades3/ | assembly_graph.fastg | metaSPAdes assembly graph in FASTG format |
| spades3/ | assembly_graph_with_scaffolds.gfa | metaSPAdes assembly graph and scaffolds paths in GFA 1.0 format |
| spades3/ | before_rr.fasta | contigs before repeat resolution |
| spades3/ | contigs.fasta | metaSPAdes resulting contigs |
| spades3/ | contigs.paths | paths in the assembly graph corresponding to contigs.fasta |
| spades3/ | dataset.info | internal configuration file |
| spades3/ | first_pe_contigs.fasta | preliminary contigs of iterative kmers assembly |
| spades3/ | input_dataset.yaml | internal YAML data set file |
| spades3/ | params.txt | information about SPAdes parameters in this run |
| spades3/ | scaffolds.fasta | metaSPAdes resulting scaffolds |
| spades3/ | scaffolds.paths | paths in the assembly graph corresponding to scaffolds.fasta |
| spades3/ | spades.log | metaSPAdes log |
| final_assembly | create_agp task result directory | |
| final_assembly/berkeleylab-jgi-meta-60ade422cd4e | directory containing checking resource script | |
| final_assembly/ | assembly.agp | an AGP format file describes the assembly |
| final_assembly/ | assembly_contigs.fna | Final assembly contig fasta |
| final_assembly/ | assembly_scaffolds.fna | Final assembly scaffolds fasta |
| final_assembly/ | assembly_scaffolds.legend | name mapping file from spades node name to new name |
| final_assembly/ | rc | cromwell script sbumit return code |
| final_assembly/ | resources.log | resource checking log |
| final_assembly/ | script | Task run commands |
| final_assembly/ | script.background | Bash script to run script.submit |
| final_assembly/ | script.submit | cromwell submit commands |
| final_assembly/ | stats.json | assembly statistics in json format |
| final_assembly/ | stderr | standard error where task writes error message to |
| final_assembly/ | stderr.background | standard error where bash script writes error message to |
| final_assembly/ | stdout | standard output where task writes error message to |
| final_assembly/ | stdout.background | standard output where bash script writes error message to |
| mapping | maps reads back to the final assembly result directory | |
| mapping/ | covstats.txt | contigs coverage informaiton |
| mapping/ | mapping_stats.txt | contigs coverage informaiton (same as covstats.txt) |
| mapping/ | pairedMapped.bam | reads mapping back to the final assembly bam file |
| mapping/ | pairedMapped.sam.gz | reads mapping back to the final assembly sam.gz file |
| mapping/ | pairedMapped_sorted.bam | reads mapping back to the final assembly sorted bam file |
| mapping/ | pairedMapped_sorted.bam.bai | reads mapping back to the final assembly sorted bam index file |
| mapping/ | rc | cromwell script sbumit return code |
| mapping/ | resources.log | resource checking log |
| mapping/ | script | Task run commands |
| mapping/ | script.background | Bash script to run script.submit |
| mapping/ | script.submit | cromwell submit commands |
| mapping/ | stderr | standard error where task writes error message to |
| mapping/ | stderr.background | standard error where bash script writes error message to |
| mapping/ | stdout | standard output where task writes error message to |
| mapping/ | stdout.background | standard output where bash script writes error message to |
Version History¶
- 1.0.1 (release date 02/16/2021; previous versions: 1.0.0)
Point of contact¶
- Original author: Brian Foster <bfoster@lbl.gov>
- Package maintainer: Chienchi Lo <chienchi@lanl.gov>