Metaproteomic Workflow (v1.0.0)¶
Summary¶
The metaproteomics workflow/pipeline is an end-to-end data processing workflow for protein identification and characterization using MS/MS data. Briefly, mass spectrometry instrument generated data files(.RAW) are converted to mzML, an open data format, using MSConvert. Peptide identification is achieved using MSGF+ and the associated metagenomic information in the FASTA (protein sequences) file format. Intensity information for identified species is extracted using MASIC and combined with protein information.
Workflow Diagram¶
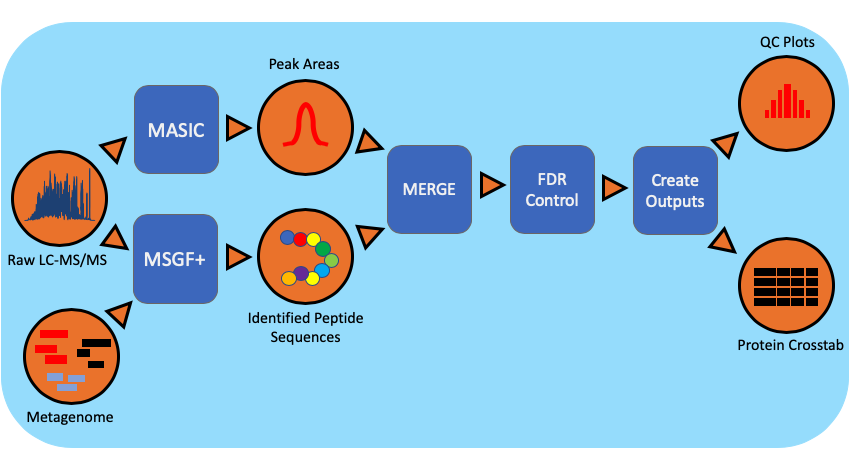
Workflow Dependencies¶
Third party software¶
|----------------------------|------------------------------------------|
| MSGFPlus | v20190628 |
| Mzid-To-Tsv-Converter | v1.3.3 |
| PeptideHitResultsProcessor | v1.5.7130 |
| pwiz-bin-windows | x86_64-vc141-release-3_0_20149_b73158966 |
| MASIC | v3.0.7235 |
| sqlite-netFx-full-source | 1.0.111.0 |
| Conda | (3-clause BSD) |
| | |
Workflow Availability¶
The workflow is available in GitHub: https://github.com/microbiomedata/metaPro
The container is available at Docker Hub (microbiomedata/mepro): https://hub.docker.com/r/microbiomedata/mepro
Inputs¶
- .raw, metagenome, parameter files : MSGFplus & MASIC, contaminant_file
Outputs¶
- Processing multiple datasets.
.
├── Data/
├── FDR_table.csv
├── Plots/
├── dataset_job_map.csv
├── peak_area_crosstab_by_dataset_id.csv
├── protein_peptide_map.csv
├── specID_table.csv
└── spectra_count_crosstab_by_dataset_id.csv
- Processing single FICUS dataset.
- metadatafile, [Example](https://jsonblob.com/400362ef-c70c-11ea-bf3d-05dfba40675b)
| Keys | Values |
|--------------------|--------------------------------------------------------------------------|
| id | str: "md5 hash of $github_url+$started_at_time+$ended_at_time" |
| name | str: "Metagenome:$proposal_extid_$sample_extid:$sequencing_project_extid |
| was_informed_by | str: "GOLD_Project_ID" |
| started_at_time | str: "metaPro start-time" |
| ended_at_time | str: "metaPro end-time" |
| type | str: tag: "nmdc:metaPro" |
| execution_resource | str: infrastructure name to run metaPro |
| git_url | str: "url to a release" |
| dataset_id | str: "dataset's unique-id at EMSL" |
| dataset_name | str: "dataset's name at EMSL" |
| has_inputs | json_obj |
| has_outputs | json_obj |
| stats | json_obj |
has_inputs :
| MSMS_out | str: file_name \|file_size \|checksum |
| metagenome_file | str: file_name \|file_size \|checksum \|
int: entry_count(#of gene sequences) \|
int: duplicate_count(#of duplicate gene sequences) |
| parameter_files | str: for_masic/for_msgfplus : file_name \|file_size \|checksum
parameter file used for peptide identification search
| Contaminant_file | str: file_name \|file_size \|checksum
(FASTA containing common contaminants in proteomics)
has_outputs:
| collapsed_fasta_file | str: file_name \|file_size \|checksum |
| resultant_file | str: file_name \|file_size \|checksum |
| data_out_table | str: file_name \|file_size \|checksum |
stats:
| from_collapsed_fasta | int: entry_count(#of unique gene sequences) |
| from_resultant_file | int: total_protein_count |
| from_data_out_table | int: PSM(# of MS/MS spectra matched to a peptide sequence at 5% false discovery rate (FDR)
float: PSM_identification_rate(# of peptide matching MS/MS spectra divided by total spectra searched (5% FDR)
int: unique_peptide_seq_count(# of unique peptide sequences observed in pipeline analysis 5% FDR)
int: first_hit_protein_count(# of proteins observed assuming single peptide-to-protein relationships)
int: mean_peptide_count(Unique peptide sequences matching to each identified protein.)
- data_out_table
| DatasetName | PeptideSequence | FirstHitProtein | SpectralCount | sum(MasicAbundance) | GeneCount | FullGeneList | FirstHitDescription | DescriptionList | min(Qvalue) |
- collapsed_fasta_file
- resultant_file
Requirements for Execution¶
- Docker or other Container Runtime
Version History¶
- 1.0.0
Point of contact¶
Package maintainer: Anubhav <anubhav@pnnl.gov>