The Read-based Taxonomy Classification (v1.0.1)¶
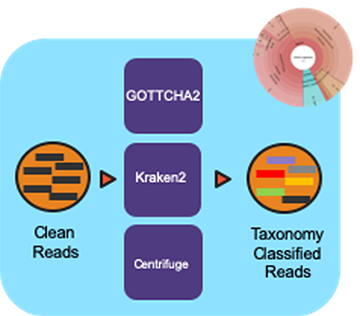
Workflow Overview¶
The pipeline takes in sequencing files (single- or paired-end) and profiles them using multiple taxonomic classification tools with the Cromwell as the workflow manager.
Workflow Availability¶
The workflow is available in GitHub: https://github.com/microbiomedata/ReadbasedAnalysis; the corresponding Docker image is available in DockerHub: https://hub.docker.com/r/microbiomedata/nmdc_taxa_profilers
Requirements for Execution:¶
(recommendations are in bold)
- WDL-capable Workflow Execution Tool (Cromwell)
- Container Runtime that can load Docker images (Docker v2.1.0.3 or higher)
Hardware Requirements:¶
- Disk space: 152 GB for databases (55 GB, 89 GB, and 8 GB for GOTTCHA2, Kraken2 and Centrifuge databases, respectively)
- 60 GB RAM
Workflow Dependencies¶
Third party software:¶
(These are included in the Docker image.)
- GOTTCHA2 v2.1.6 (License: BSD-3-Clause-LANL)
- Kraken2 v2.0.8 (License: MIT)
- Centrifuge v1.0.4 (License: GPL-3)
Requisite databases:¶
The database for each tool must be downloaded and installed. These databases total 152 GB.
- GOTTCHA2 database (gottcha2/):
The database RefSeqr90.cg.BacteriaArchaeaViruses.species.fna contains complete genomes of bacteria, archaea and viruses from RefSeq Release 90. The following commands will download the database:
wget https://edge-dl.lanl.gov/GOTTCHA2/RefSeq-r90.cg.BacteriaArchaeaViruses.species.tar
tar -xvf RefSeq-r90.cg.BacteriaArchaeaViruses.species.tar
rm RefSeq-r90.cg.BacteriaArchaeaViruses.species.tar
- Kraken2 database (kraken2/):
This is a standard Kraken 2 database, built from NCBI RefSeq genomes. The following commands will download the database:
mkdir kraken2
wget https://genome-idx.s3.amazonaws.com/kraken/k2_standard_20201202.tar.gz
tar -xzvf k2_standard_20201202.tar.gz -C kraken2
rm k2_standard_20201202.tar.gz
- Centrifuge database (centrifuge/):
This is a compressed database built from RefSeq genomes of Bacteria and Archaea. The following commands will download the database:
mkdir centrifuge
wget https://genome-idx.s3.amazonaws.com/centrifuge/p_compressed_2018_4_15.tar.gz
tar -xzvf p_compressed_2018_4_15.tar.gz -C centrifuge
rm p_compressed_2018_4_15.tar.gz
Sample dataset(s):¶
Zymobiomics mock-community DNA control (SRR7877884); this dataset is ~7 GB.
Input: A JSON file containing the following information: 1. selection of profiling tools (set as true if selected) 2. the paths to the required database(s) for the tools selected 3. the paths to the input fastq file(s) (paired-end data is shown; this can be the output of the Reads QC workflow in interleaved format which will be treated as single-end data.) 4. the prefix for the output file names 5. the path of the output directory 6. CPU number requested for the run.
{
"ReadbasedAnalysis.enabled_tools": {
"gottcha2": true,
"kraken2": true,
"centrifuge": true
},
"ReadbasedAnalysis.db": {
"gottcha2": "/path/to/database/RefSeq-r90.cg.BacteriaArchaeaViruses.species.fna",
"kraken2": " /path/to/kraken2",
"centrifuge": "/path/to/centrifuge/p_compressed"
},
"ReadbasedAnalysis.reads": [
"/path/to/SRR7877884.1.fastq.gz",
"/path/to/SRR7877884.2.fastq.gz"
],
"ReadbasedAnalysis.paired": true,
"ReadbasedAnalysis.prefix": "SRR7877884",
"ReadbasedAnalysis.outdir": "/path/to/ReadbasedAnalysis",
"ReadbasedAnalysis.cpu": 4
}
Output:¶
The workflow creates an output JSON file and individual output sub-directories for each tool which include tabular classification results, a tabular report, and a Krona plot (html).:
ReadbasedAnalysis/
|-- SRR7877884.json
|-- centrifuge
| |-- SRR7877884.classification.tsv
| |-- SRR7877884.report.tsv
| `-- SRR7877884.krona.html
|
|-- gottcha2
| |-- SRR7877884.full.tsv
| |-- SRR7877884.krona.html
| `-- SRR7877884.tsv
|
`-- kraken2
|-- SRR7877884.classification.tsv
|-- SRR7877884.krona.html
`-- SRR7877884.report.tsv
Below is an example of the output directory files with descriptions to the right.
| FileName | Description |
| SRR7877884.json | ReadbasedAnalysis result JSON file |
| centrifuge/SRR7877884.classification.tsv | Centrifuge output read classification TSV file |
| centrifuge/SRR7877884.report.tsv | Centrifuge output report TSV file |
| centrifuge/SRR7877884.krona.html | Centrifuge krona plot HTML file |
| gottcha2/SRR7877884.full.tsv | GOTTCHA2 detail output TSV file |
| gottcha2/SRR7877884.tsv | GOTTCHA2 output report TSV file |
| gottcha2/SRR7877884.krona.html | GOTTCHA2 krona plot HTML file |
| kraken2/SRR7877884.classification.tsv | Kraken2 output read classification TSV file |
| kraken2/SRR7877884.report.tsv | Kraken2 output report TSV file |
| kraken2/SRR7877884.krona.html | Kraken2 krona plot HTML file |
Version History¶
1.0.1 (release date 01/14/2021; previous versions: 1.0.0)
Point of contact¶
Package maintainer: Po-E Li <po-e@lanl.gov>